Darwin: a genomics co-processor provides up to 15,000x acceleration on long read assembly Turakhia et al., ASPLOS’18
With the slow demise of Moore’s law, hardware accelerators are needed to meet the rapidly growing computational requirements of X.
For this paper, X = genomics, and genomic data is certainly growing fast: doubling every 7 months and on track to surpass YouTube and Twitter by 2025. Rack-size machines can sequence 50 genomes a day, portable sequencers require several days per genome. Third-generation sequencing technologies are now available which produce much longer reads of contiguous DNA – on the order of tens of kilobases compared to only a few hundred bases with the previous generations of technology.
For personalized medicine, long reads are superior in identifying structural variants i.e. large insertions, deletions and re-arrangements in the genome spanning kilobases or more which are sometimes associated with diseases; for haplotype phasing, to distinguish mutations on maternal vs paternal chromosomes; and for resolving highly repetitive regions in the genome.
The long read technology comes with a drawback though – high error rates in sequencing of between 15%-40%. The errors are corrected using computational methods ‘that can be orders of magnitude slower than those used for the second generation technologies.’
This paper describes “Darwin”— a co-processor for genomic sequence alignment that , without sacrificing sensitivity, provides up to 15,000x speedup over the state-of-the-art software for reference-guided assembly of third-generation reads. Darwin achieves this speedup through hardware/algorithm co-design…
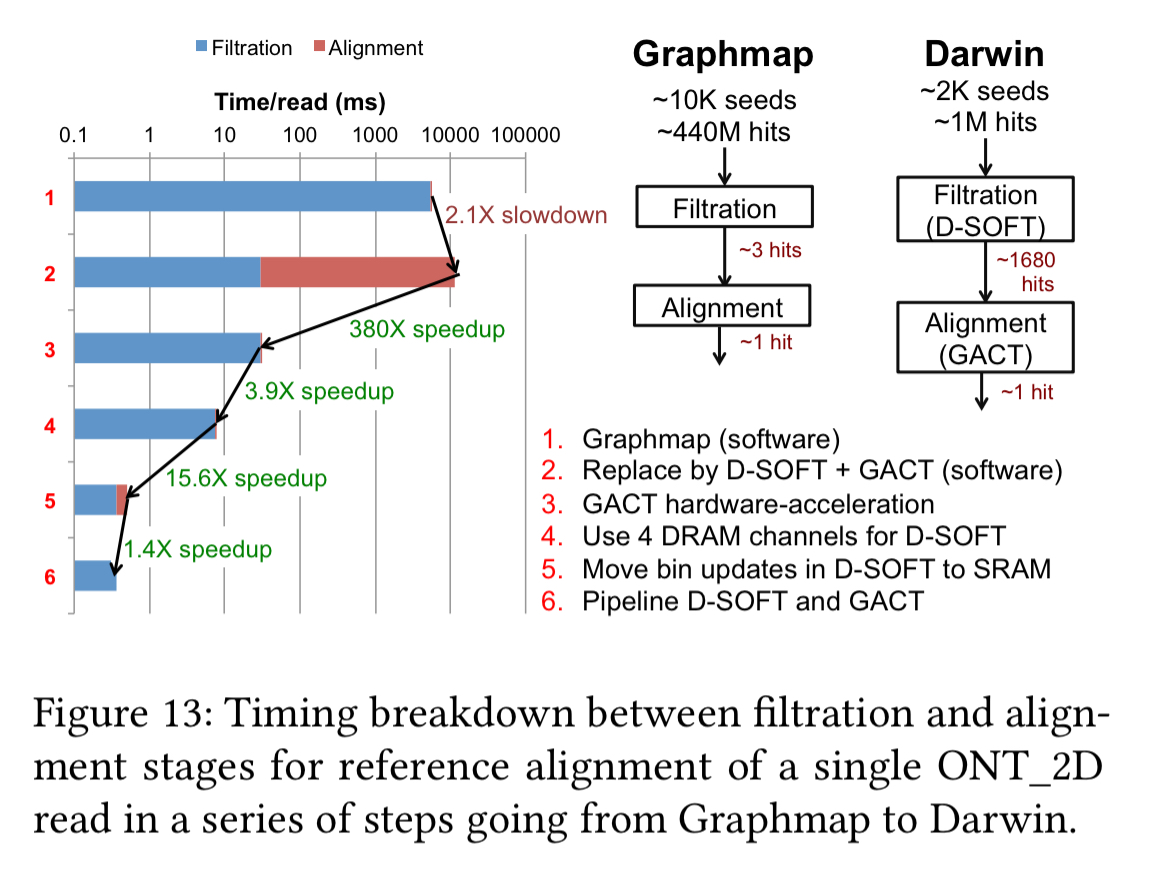
The algorithms are D-SOFT and GACT. D-SOFT (Diagonal-band Seed Overlapping based Filtration Technique) implements the filtering (seeding) stage of alignment. GACT (Genome Alignment using Constant memory Traceback) uses dynamic programming for aligning arbitrarily long sequences using constant memory for the compute intensive step. Both algorithms have accompanying hardware accelerators, which are prototyped using FPGAs and also embodied in an ASIC design.
Genome sequence alignment and assembly
The sequence alignment problem takes a query sequence Q of letters in 
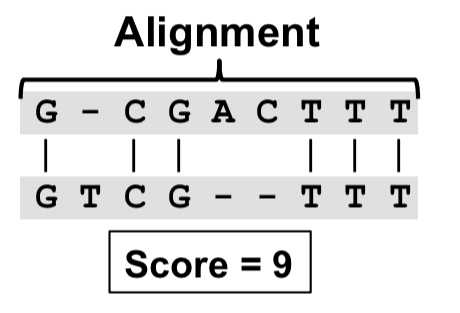
The seed-and-extend approach starts with substrings (the seeds) of fixed size k drawn from Q, and finds their exact matches in R (called seed hits). Once seed hits are found, the cells surrounding the hits are explored (using a dynamic programming approach). This avoids the high cost of exploring the full space. A drawback of the seed-and-extend approach is that alignments with no exactly matching substrings of length greater than or equal to k will not be discovered, reducing the algorithm’s sensitivity.
Sequencing is the process of shearing DNA molecules into a large number N of random fragments and reading the bases in each fragment. N is chosen such that each base-pair is expected to be covered multiple times, enabling errors to be corrected via a consensus mechanism.
Genome assembly is the process of reconstructing a genome G from a set of reads S. The reconstruction can be reference guided, using reference sequence R to guide re-assembly, or it can be de novo, reconstructing G from S without the benefit (and bias!) of R.
D-SOFT filtering
For long reads with high error rates, simple seeding algorithms like BLAST give a high false positive rate that results in excessive computation in the extend or alignment stage. To address this issue, Myers14 uses multiple seeds from Q, counting the number of matching bases in a band of diagonals and then filtering on a fixed threshold.
D-SOFT uses a similar filtering strategy, but with a different implementation.
Algorithm
D-SOFT filters by counting the number of unique bases in Q covered by seed hits in diagonal bands. If the count in a given band exceeds h bases, then the band is selected for alignment.
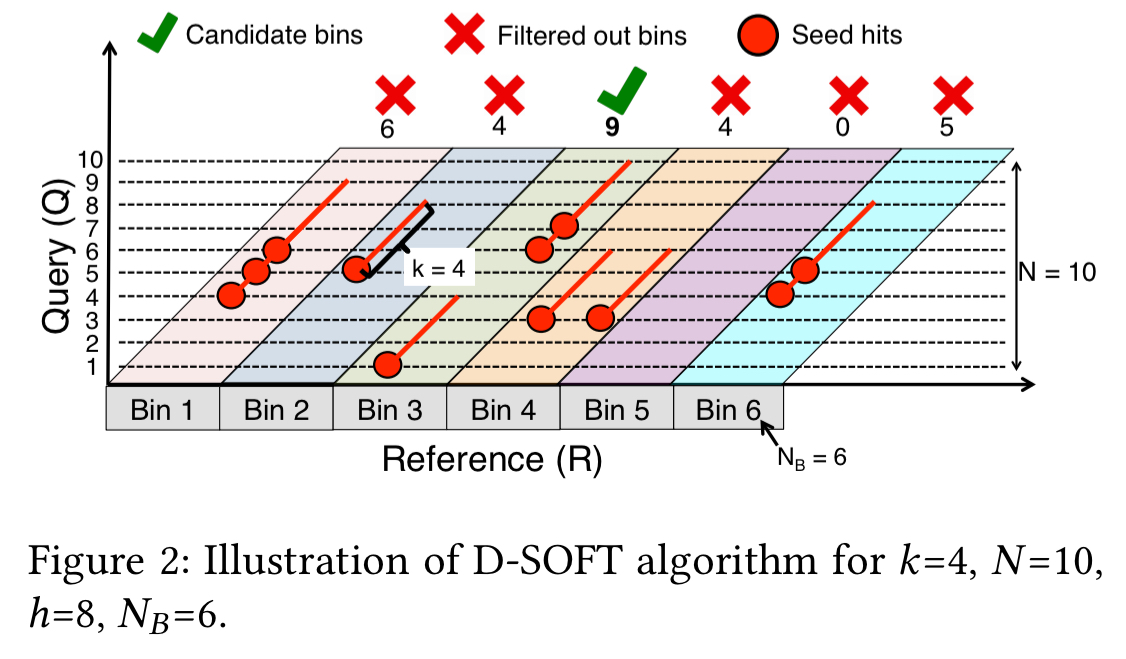
In the figure above, the X axis corresponds to positions in R, divided into 
The count for a diagonal is then simply the number of bases in Q covered by seed hits in that band. In the example above, both Bin1 and Bin 3 have three seed hits, but in Bin 1 only 6 bases are covered, whereas in Bin 3 there are 9 covered bases. With the threshold h set to 8, only bin 3 will be chosen as a candidate.
Counting unique bases in a diagonal band allows D-SOFT to be more precise at high sensitivity than strategies that count seed hits, because overlapping bases are not counted multiple times.
Seed lookup is implemented using a seed position table: for each of the 
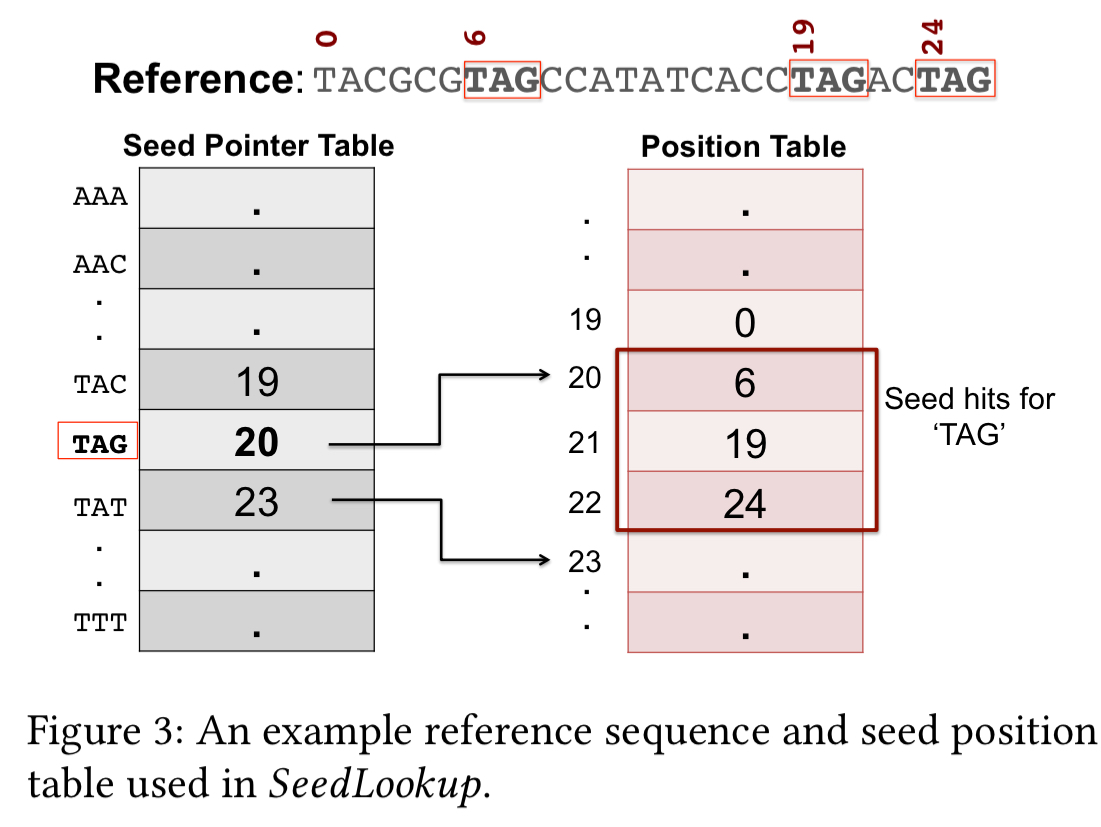
Accelerator design
The D-SOFT accelerators maps the sequentially accessed seed tables to off-chip DRAM and places the randomly-accessed bin arrays in banks of on-chip SRAM. Incoming seeds are distributed over four DRAM channels, where a seed-position lookup block looks up the hits in the position table and calculates the bin of each hit. Bin-offset pairs are then sent via a network-on-chip to the on-chip SRAM bank associated with the bin.
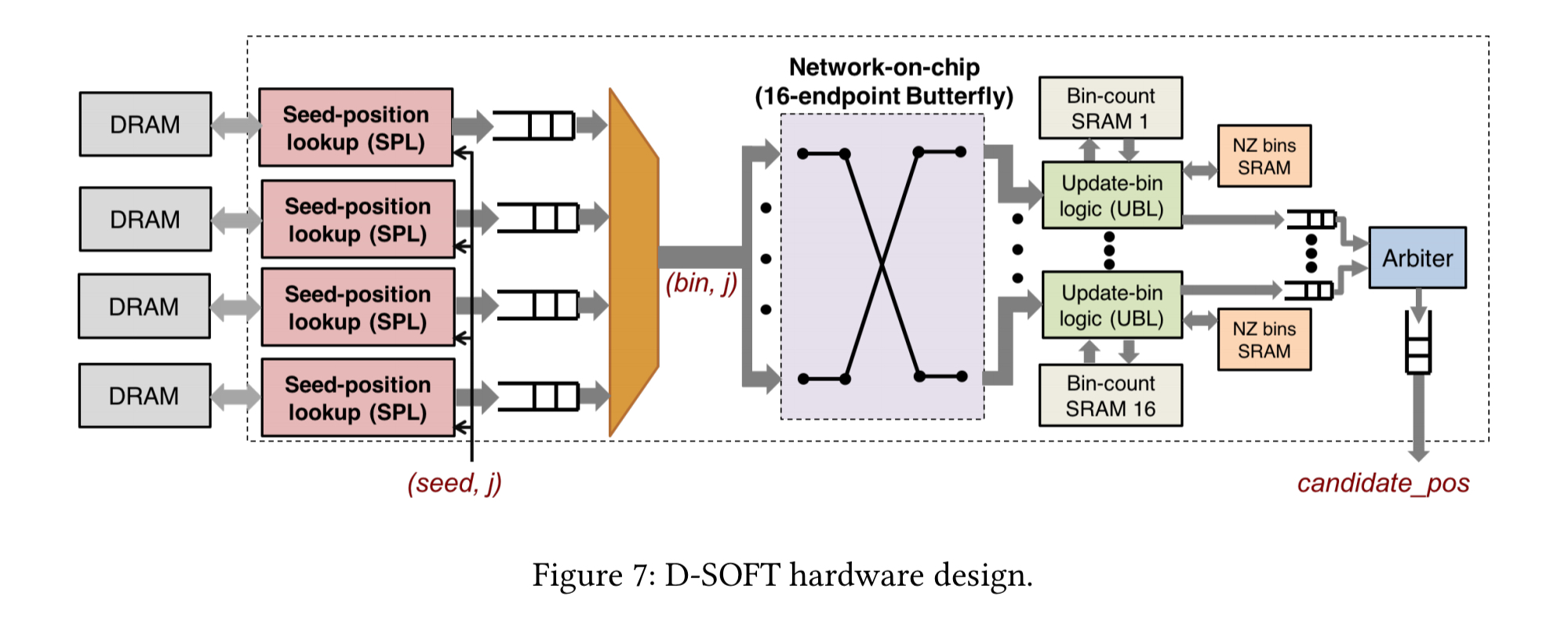
With 64MB SRAMs divided into 16 banks, up to 32M bins can accommodate at 4Gbp reference sequence with B=128.
GACT alignment
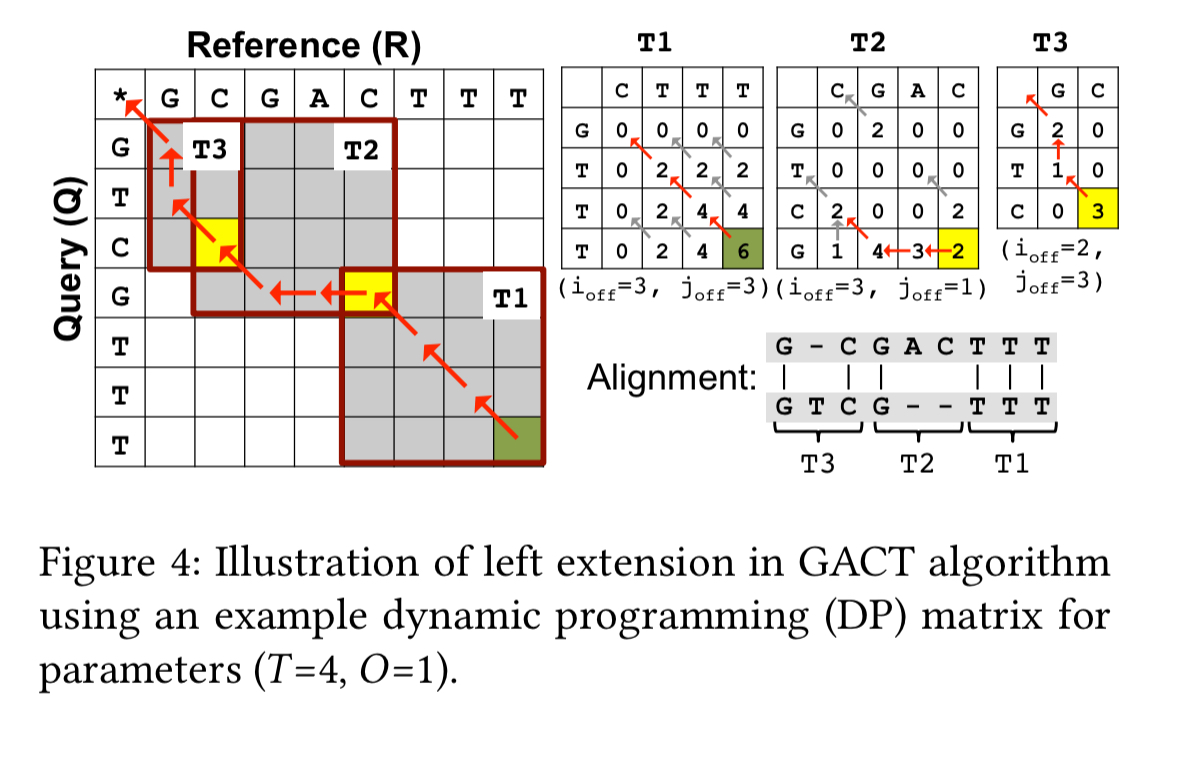
GACT performs the alignment extension part of a seed-and-extend system. It finds an arbitrarily long alignment extension in linear time by following the optimal path within overlapping tiles. GACT uses two parameters, the tile size T and an overlap threshold O (less than T). Empirically, we have found that GACT gives an optimal result (identical to Smith-Waterman) with reasonable values of T and Q.
Algorithm
The algorithm for extending sequences to the left is show below, reverse R and Q for extending to the right:

Accelerator design
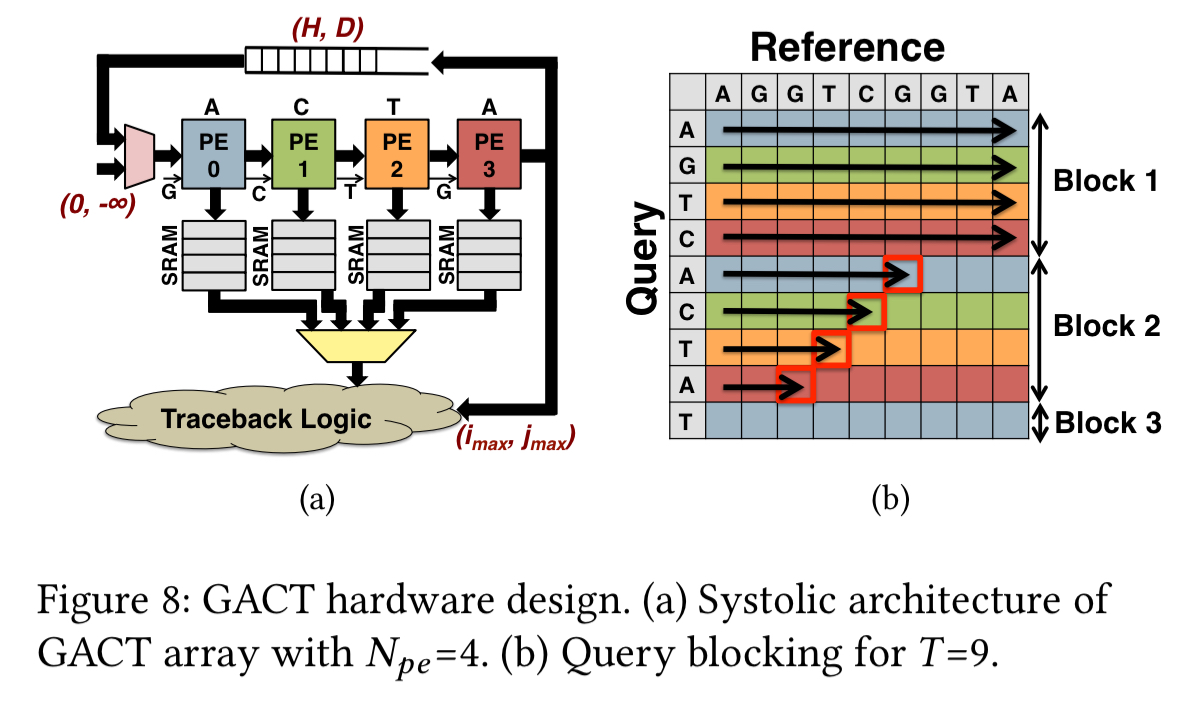
The GACT array accelerators handle the compute-intensive align routine in GACT, with the rest of the algorithm implemented in software.
A linear array of
processing elements (PEs) exploits wavefront parallelism to compute up to
The three scores are the affine gap penalties for insertion and deletion, and the overall cell score.
Darwin: putting it all together
The Darwin co-processor combines the hardware-accelerated GACT and D-SOFT algorithms. It uses four 32GB LPDDR4 DRAM memory channels, storing identical copies of the seed position table, the reference sequence, and the query sequences. This helps to keep all channels load-balanced and fully utilised at all times.
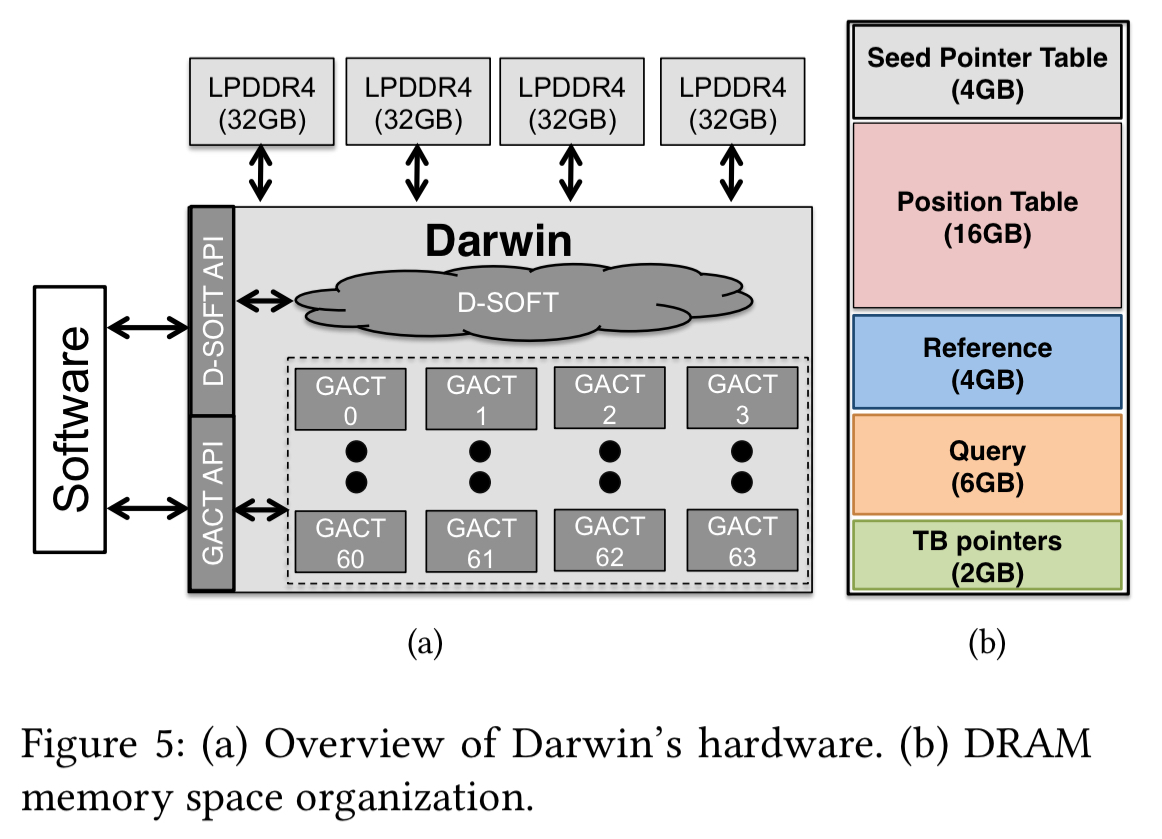
For both reference guided and de novo assembly the forward and reverse complement of P reads are used as queries into a reference sequence R. For reference guided assembly an actual reference sequence is used. For de novo assembly the reads are concatenated to form the reference sequence, with each read padded to take up a whole number of bins. Candidate bins from D-SOFT are passed to the GACT array which extends and scores the alignment.

The assembly algorithms are partitioned between hardware and software as follows. Software initializes D-SOFT and GACT parameters and receives candidate alignment positions from D-SOFT, which is accelerated in hardware. Align – the compute extensive step in GACT – is hardware accelerated, the remainder of the alignment step is performed in software. Software also constructs the seed position table in de novo assembly.
Evaluation
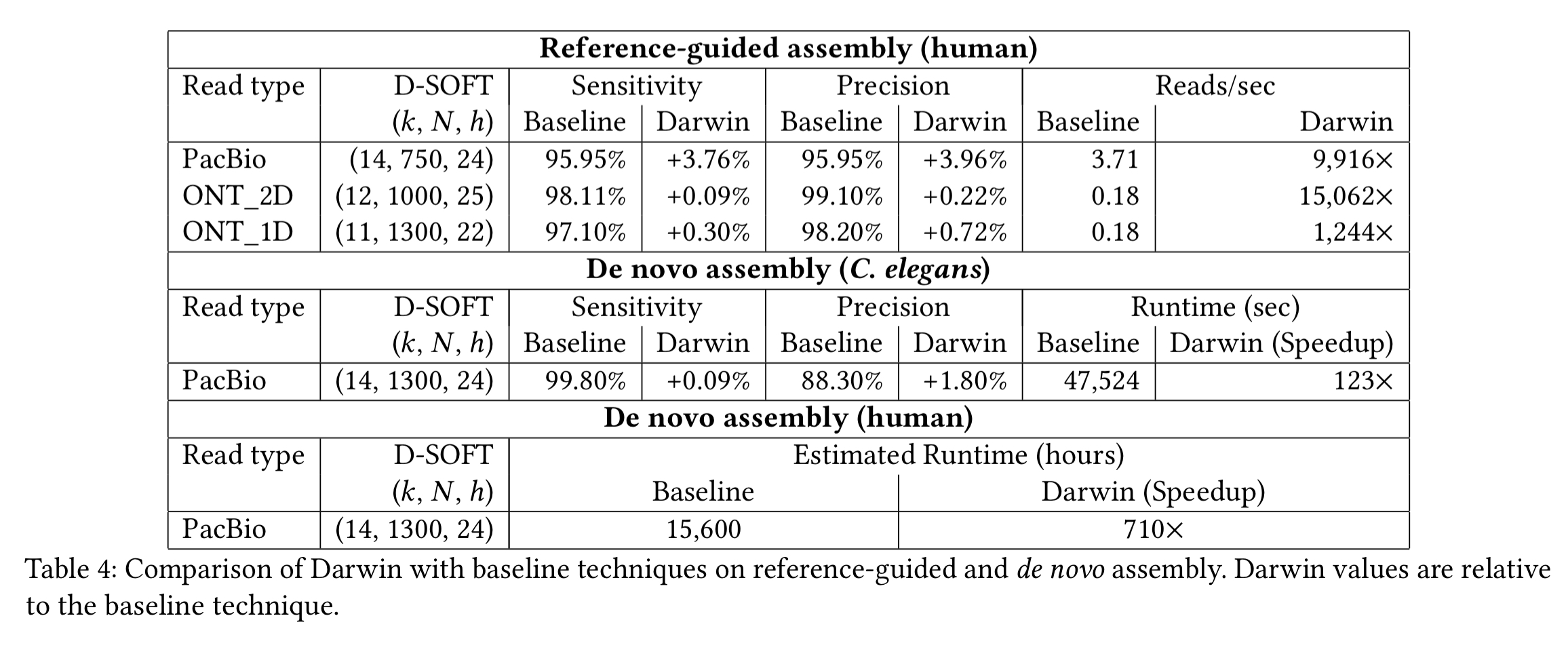
The following table compares Darwin’s performance against baseline algorithms for reference-guided assembly of the human genome using 50,000 randomly sampled reads for each of three read sets, as well as de novo assembly of a C. elegans genome and a human genome. The D-SOFT parameters in Darwin are adjusted to match or exceed the sensitivity of the baseline technique. Darwin achieves speedups of up to 15,000x!
The following chart (log scale) shows the breakdown of where all those performance gains are coming from.
ASIC synthesis was carried out using the Synopsys Design Compiler in a TSMC 40nm CMOS process. The area and power breakdown of the different components of the ASIC are shown in this table:
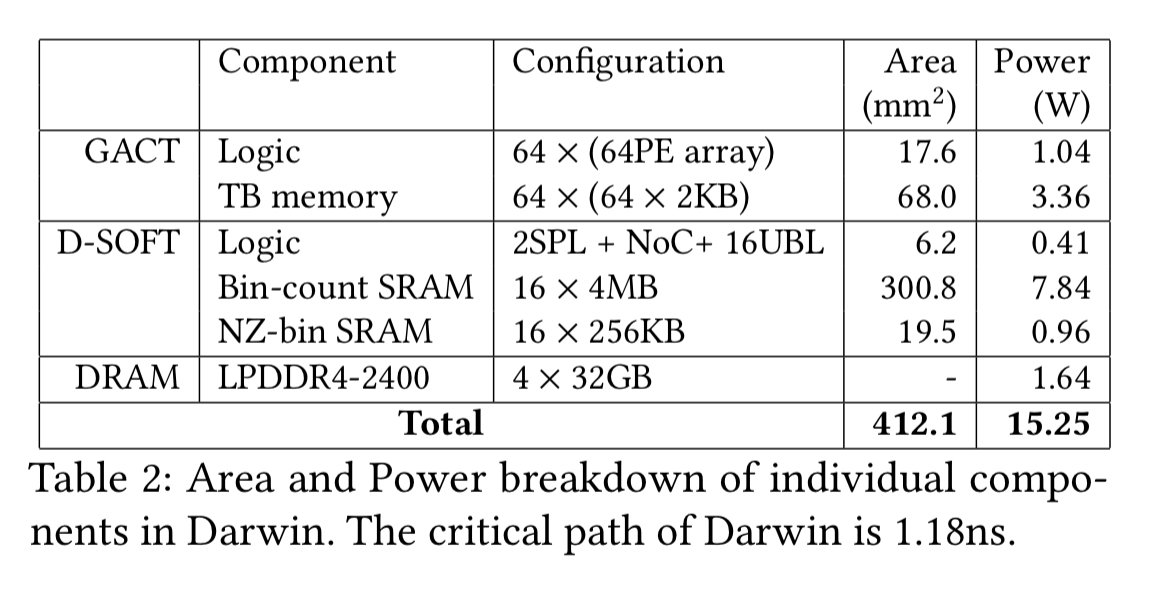
Darwin can operate at 847MHz (3.4x the FPGA frequency) with relatively small power consumption (about 15W). In a modern 14nm process Darwin would be much smaller (about 50mm squared vs 412mm squared) and consume even less power (about 6.4W).
Darwin’s filtering and alignment operations are general and can be applied to rapidly growing sequence alignment applications beyond read assembly, such as whole genome alignments, metagenomics and multiple sequence alignments.

7 thoughts on “Darwin: a genomics co-processor provides up to 15,000x acceleration on long read assembly”
Comments are closed.