A DNA-Based Archival Storage System – Bornholt et al. ASPLOS ’16
It’s pretty cool that a paper on DNA-based storage makes a conference such as ASPLOS. And as you’ll see, there are good reasons we should be taking it very seriously indeed. DNA has some very interesting properties – it’s extremely dense (1 exabyte (109 GB) per mm3) and lasts a long-time (observed. half-life of over 500 years. It doesn’t work quite like this, but to put that density into context, imagine how many mm3 of raw storage you could fit into a USB-sized form factor. I’ll guess at least 100 – that would be 100,000 PB of storage in your thumb-drive!
In this paper the authors build a DNA-based key-value store supporting random access and are successfully able to store and retrieve data. Crucially, it seems that the internet’s cute kitten image archive can be safely preserved for posterity. Here’s a test image that was synthesized and then subsequently sequenced – it’s the DNA of a cat, but not as we normally think of it!
The idea of course is not to build thumb-drives, but to create a dense and durable layer of archival storage that can cope with the volumes of data we now generate.
The “digital universe” (all digital data worldwide) is forecast to grow to over 16 zettabytes in 2017. Alarmingy, the exponential growth rate easily exceeds our ability to store it, even when accounting for forecast improvements in storage technologies. A significant fraction of this data is in archival form; for example, Facebook recently built an entire data center dedicated to 1 exabyte of cold storage.
(Recall, 1mm3 of DNA has the same raw storage potential as that entire data center!).
Storing data using DNA means first mapping digital data into DNA nucleotide sequences, synthesizing (manufacturing) the corresponding DNA molecules, and then storing them away. To read the data you sequence the DNA molecules and decode the data back to the original digital data. Synthesis and sequencing cost and latency have been dropping on an exponential curve – growth in sequencing productivity outstrips Moore’s law, and is tracked by Carlson’s Curves. Orders of magnitude in cost reduction and efficiency improvements are still expected.
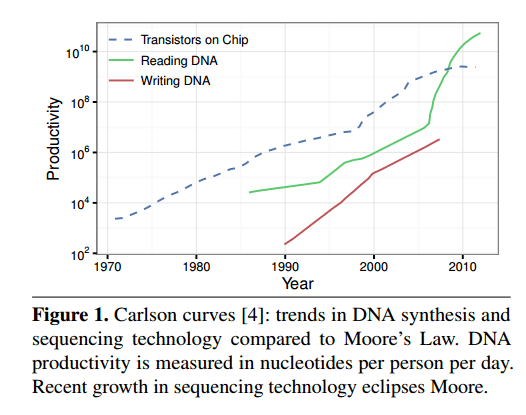
We think the time is ripe to consider DNA-based storage seriously and explore system designs and architectural implications. This paper presents an architecture for a DNA-backed archival storage system, modeled as a key-value store. A DNA storage system must overcome several challenges:
- DNA synthesis and sequencing has error rates on the order of 1% per nucleotide. Therefore appropriate encoding schemes that are tolerant to errors must be used.
- Random access can be problematic – existing work provides only large block access resulting in large read latency. The authors design a system using a polymerase chain reaction (PCR) to accelerate reads and avoid the need to sequence an entire DNA pool.
A DNA primer
DNA is comprised of four different types of nucleotides, adenine (A), cytosine (C), guanine (G), and thymine (T). A DNA strand is a linear sequence of these. Polymerase chain reaction (PCR) is a method for exponentially amplifying the concentration of selected sequences of DNA within a pool.
A PCR reaction requires four main components: the template, sequencing primers, a thermostable polymerase and individual nucleotides that get incorporated into the DNA strand being amplified. The template is a single- or double-stranded molecule containing the (sub)sequence that will be amplified. The DNA sequencing primers are short synthetic strands that define the beginning and end of the region to be amplified. The polymerase is an enzyme that creates double-stranded DNA from a single-stranded template by “filling in” individual complementary nucleotides one by one, starting from a primer bound to that template. PCR happens in “cycles”, each of which doubles the number of templates in a solution. The process can be repeated until the desired number of copies is created.
Single-strand DNA sequences can be synthesized chemically, nucleotide by nucleotide. The practical limit on sequence length is about 200 nucleotides.
In practice, synthesis of a given sequence uses a large number of parallel start sites and results in many truncated byproducts (the dominant error in DNA synthesis), in addition to many copies of the full-length target sequence. Thus, despite errors in synthesizing any specific strand, a given synthesis batch will usually produce many perfect strands.
The most popular sequencing techniques also exploit synthesis:
The strand of interest serves as a template for the polymerase, which creates a complement of the strand. Importantly, fluorescent nucleotides are used during this synthesis process. Since each type of fluorescent nucleotide emits a different color, it is possible to read out the complement sequence optically. Sequencing is error-prone, but as with synthesis, in aggregate, sequencing typically produces enough precise reads of each strand.
DNA-based encoding
Since we have four code letters (A,C,G, and T) the obvious approach to storing binary data is to encode it in base 4 with e.g. 0=A, 1=C, 2=G, and 3=T. Since synthesis and sequencing is error prone though, we need to use a more robust encoding.
The likelihood of some forms of error can be reducedby encoding binary data in base 3 instead of base 4 , as Figure 5(a) [below] illustrates. Each ternary digit maps to a DNA nucleotide based on a rotating code (Figure 5(b)) that avoids repeating the same nucleotide twice. This encoding avoids homopolymers—repetitions of the same nucleotide that significantly increase the chance of sequencing errors.
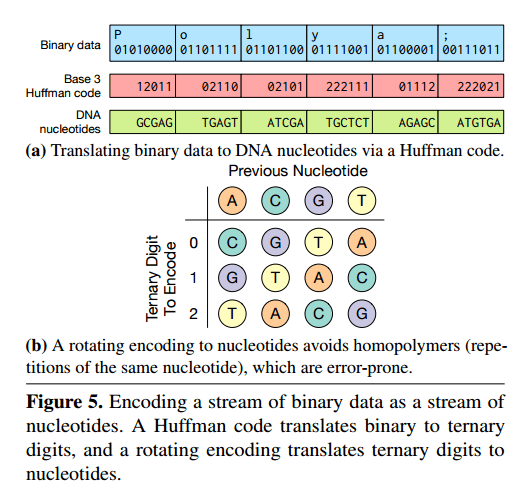
The Huffman code is used since otherwise mapping between base 2 (original data) and base 3 would be inefficient.
Since we can only generate sequences with low hundreds of nucleotides, we need to use multiple strands to hold data beyond a few hundred bits. The input stream is divided into multiple strands, each sandwiching source data between two primer sequences. These primers allow the read process to isolate molecules of interest and thus perform random access.
To provide random access, we instead design a mapping from keys to unique primer sequences. All strands for a particular object share a common primere, and different strands with the same primer are distinguised by their different addresses. Primers allow random access via a polymerase chain reaction (PCR), which produces many copies of a piece of DNA in a solution. By controlling the sequences used as primers for PCR, we can dictate which strands in the solution are amplified. To read a particular key’s value from the solution, we simply perform a PCR process using that key’s primer, which amplifies the selected strands.
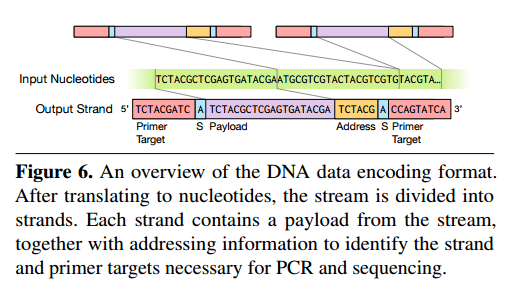
The sense nucleotides (“S” in the figure above) indicate whether the strand has been reverse complemented – which is done to avoid certain pathological cases.
Encoding data using Huffman codes and base 3 means that each bit of data is encoded in exactly one location in the output DNA strands. The authors add a further layer of redundancy at the data encoding level. Given payloads A and B, they also generate and store the strand A XOR B. Recovering any two of the three strands is then sufficient to recover the third.
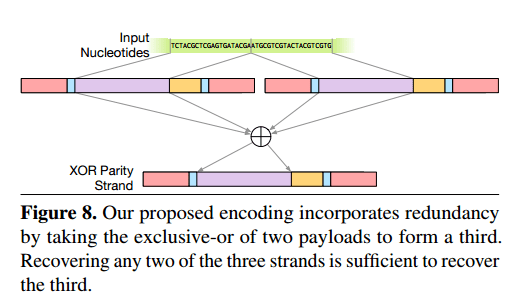
The address block of the new strand encodes the addresses of the input strands that were the inputs to the exclusive-or; the high bit of the address is used to indicate whether a strand is an original payload or an exclusive-or strand. This encoding provides its redundancy in a similar fashion to RAID 5: any two of the three strands A, B, and A ⊕ B are sufficient to recover the third.
A further trick is to compute A ⊕ B’, where B’ is B in reverse. “Since nucleotides at the end of a strand are more error-prose, reversing one strand ensures that the average quality is (roughly) constant along the strand, so that each nucleotide has at least some redundancy information stored in a high-reliability position.”
An end-to-end DNA storage system
A DNA storage system consists of a DNA synthesizer that encodes the data to be stored in DNA, a storage container with compartments that store pools of DNA that map to a volume, and a DNA sequencer that reads DNA sequences and converts them back into digital data.
Since strands can store roughly 50-100 bits total, a typical data object maps to a very large number of DNA strands. These DNA strands are stored in pools with stochastic spatial organization – thus the need to embed address information within the strands themselves using the primers.
Separating the DNA strands into a collection of pools balances a trade-off between storage density, reliability, and performance. The most dense way to store data would be to have all strands in a single pool, but this arrangement sacrifices reliability for two reasons. First, a single pool requires many different primers to distinguish all keys, which increases the chance that two primers react poorly to each other. Second, a single pool reduces the likelihood that a random sample drawn during the read process will contain all the desired data. On the other hand, using a separate pool per key sacrifices density excessively. We therefore use a library of reasonably-sized pools, and use random access within each pool.
The overall process for reading and writing is summarized in the figure below:
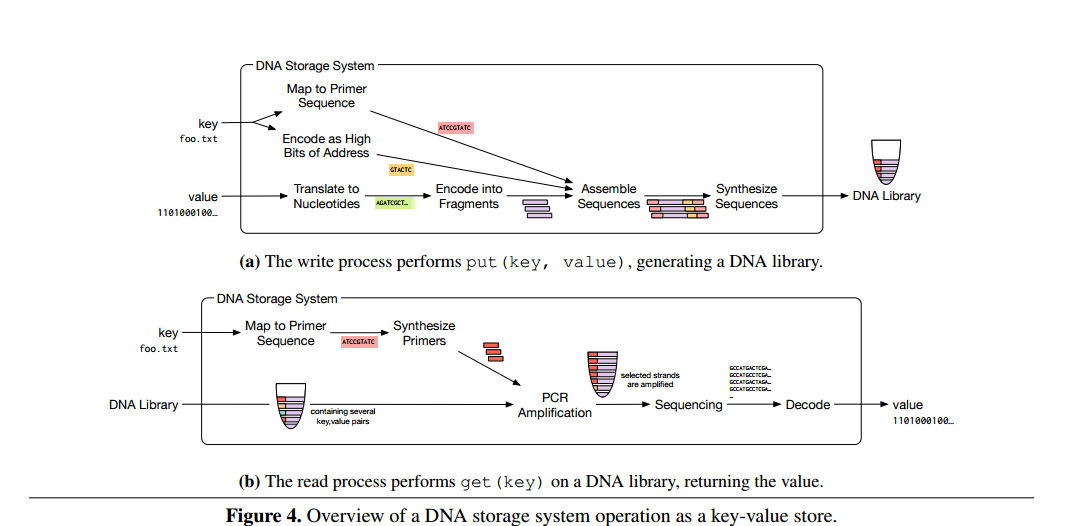
(Click to enlarge)
The read process removes a sample of DNA from the pool, and so cumulative reads reduce the quantity of DNA available for future operations. But DNA is easy to replicate, and so the pools can easily be replenished after read operations if necessary. If successive amplification is problematic, a pool can also be completely resynthesized after a read operation.
Related work
Concurrent with our work, Yazdi et al. developed a method for rewritable random-access DNA-based storage. Its encoding is dictionary-based and focuses on storage of text, while our approach accommodates arbitrary binary data. We do not support rewritability, which adds substantial complexity, because write-once is appropriate for archival items (e.g., photos) that are unlikely to change. If necessary, one can use a log-based approach for rewriting objects, since the density of DNA exceeds the overhead of logging.

Reblogged this on Collected Links.